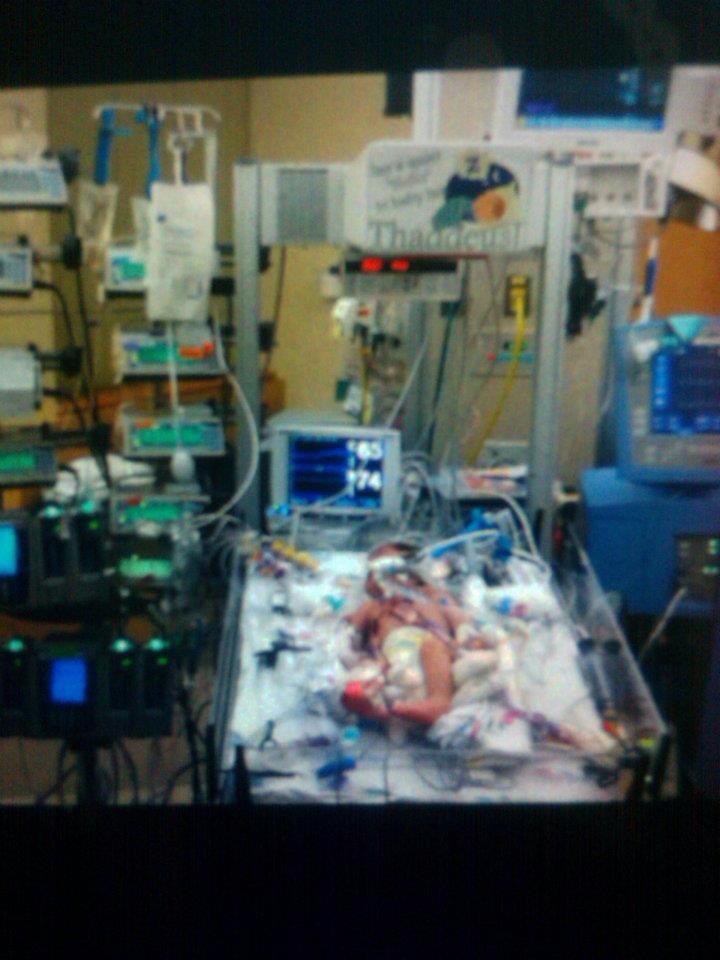
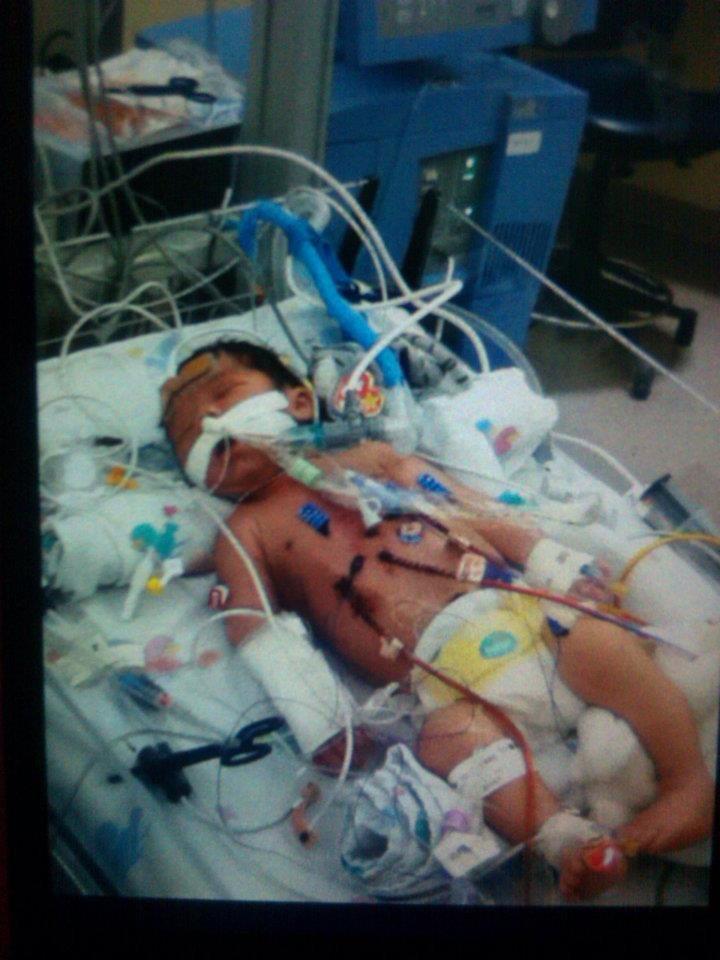
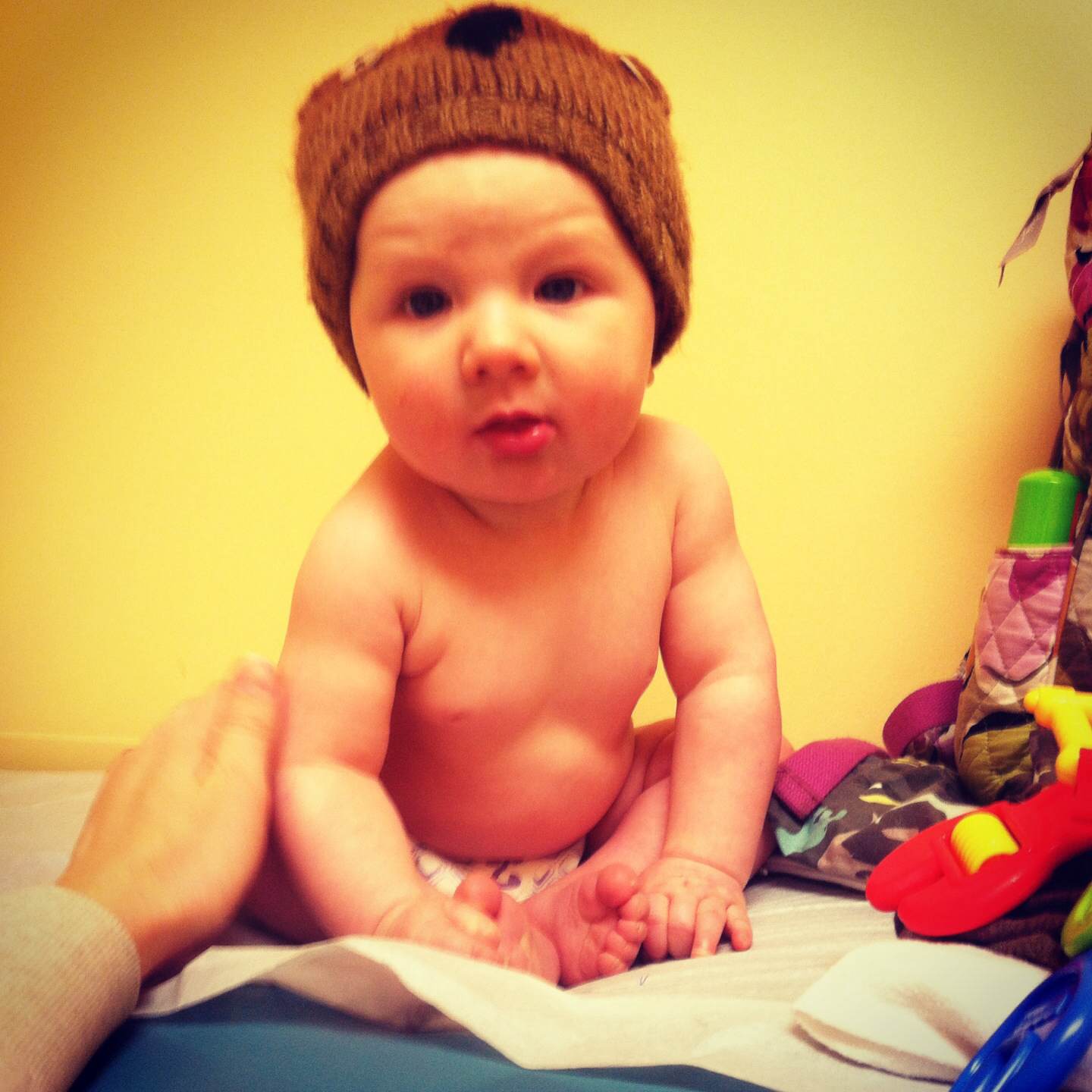
It was a happy coincidence that the story I shared on Friday was about a child who was born with a congenital heart defect. Congenital Heart Defect Awareness Week is Feb. 7-14. I didn’t think about that when I chose Thaddeus’s story for Friday, but I’m happy it worked out that way.
I am going to share stories on Wednesday and Friday of two other children born with congenital heart defects. However, today I plan to share Lucy’s story, since her birthday is tomorrow. She was born two years ago tonight, at 12:15 a.m. Since I am Lucy’s mom, this story is definitely in my own words. J
As far as we know, Lucy’s heart was fine during my pregnancy and when she was born. We don’t know exactly when her heart started to go bad or what caused it. The doctors were certain that she had dilated cardiomyopathy for quite some time before we discovered it, but they did not think she was born with it. Her heart did not get in the condition it was in overnight.
On Nov. 4, 2012, Chris and I both noticed that Lucy was breathing fast. That evening when I put her to bed, I felt inexplicably worried about her. As I held her outside of her bedroom door that evening, I thought I saw a dark figure standing in her dark room. It was enough to make me do a double-take. I now think it was my shadow on her closet door, but it seems like a sign regardless, in light of what happened later.
Chris reported after he checked on her that night that she was still breathing really fast. Chris is the less likely to worry of the two of us, so I took that as a sign that this warranted looking into.
I made an appointment with our pediatrician the next morning. I noticed that she was retracting when she breathed. I remembered Max doing that when he had RSV at six months. I figured that’s what she had, too, and the sequence of events bore out that conclusion at first.
The pediatrician ordered an x-ray. We ended up waiting at the office so long that Chris picked Max up from school and brought him up there. The doctor said her lungs looked streaky on the x-ray and prescribed nebulizer treatments, just like Max had. We did one in the office. We discussed who would go pick up the nebulizer equipment and wondered whether we still had Max’s at home.
We were in the car about to leave, when my phone rang. The doc wanted us to come back in for a blood test. I can’t remember if he and the nurse told us at the time or if we could just tell that they were troubled. Something wasn’t adding up about the way she looked and the way she was breathing. She looked perfectly healthy, but she was breathing so fast.
We had the blood test and went back to an exam room to await the results. Dr. P finally came back in, looking troubled. He told us that he’d finally gotten a look at her x-rays and her heart was enlarged. He was sending us to a pediatric cardiologist for an Echo. I can’t remember what else he told us, but we left perplexed, but not too worried yet. I think he mentioned something about medicine possibly fixing the condition.
We arrived at the cardiologist’s office and they did an Echo. Then, we returned to an exam room and waited. And waited.
I finally wore down. We had been shunted from one test to another and one exam room to another all day. I began to sob, telling Chris that something was really wrong or the doctor would have come in by now. I was terrified.
She finally came in and told us that Lucy had dilated cardiomyopathy. She mentioned that and viral myocarditis, I think. For the next few days, we really weren’t sure which one Lucy had. She also said that there were medicines that could help and Lucy had to be hospitalized. She said we could drive her to Dell Children’s ourselves or we could go in an ambulance.
We chose to drive her ourselves. I called my parents on the way and told them we needed them to come down. I remember Chris’s frustration at the traffic and his desperation to get her to the hospital. For once, I was the calm one and I reminded him that they wouldn’t have let us take her if it was imperative she get there right away.
We arrived and were quickly ushered back into the ER. No waiting room for us that time. Another cardiologist from the same practice was the first to utter the “T” word: transplant. Chris and I were flabbergasted. We both walked around the exam room crying, while Max ate the chicken strips a nurse kindly brought and Lucy babbled and played with toys on the bed. We both thought our daughter was going to die.
We became increasingly agitated the longer we waited in the ER. If there was medicine that could help her, why weren’t they hurrying to start it? It drove me mad that my daughter apparently had a life-threatening condition and no one was rushing to get her to a room and start her on medication.
We finally got back to a room. I chose to stay the first night and Chris and Max headed home at about 11 pm. Our journey had begun.
I have several “snapshots” in my head of certain moments that day. I remember the way Lucy looked sitting on the living room rug, playing with a toy while I watched the Halloween special of “Pretty Little Liars”. I remember the gray onesie with pink edging she had on.
I remember driving on the flyover from 183 to Mopac, posting on Facebook about Lucy’s heart being enlarged and asking for prayers. I remember that my friend Lea was the first to comment.
I remember sitting in the wheelchair with Lucy as they moved her from the ER to her room in the PICU. I clutched her so tightly, hunching my entire body around her, trying to protect both of us.
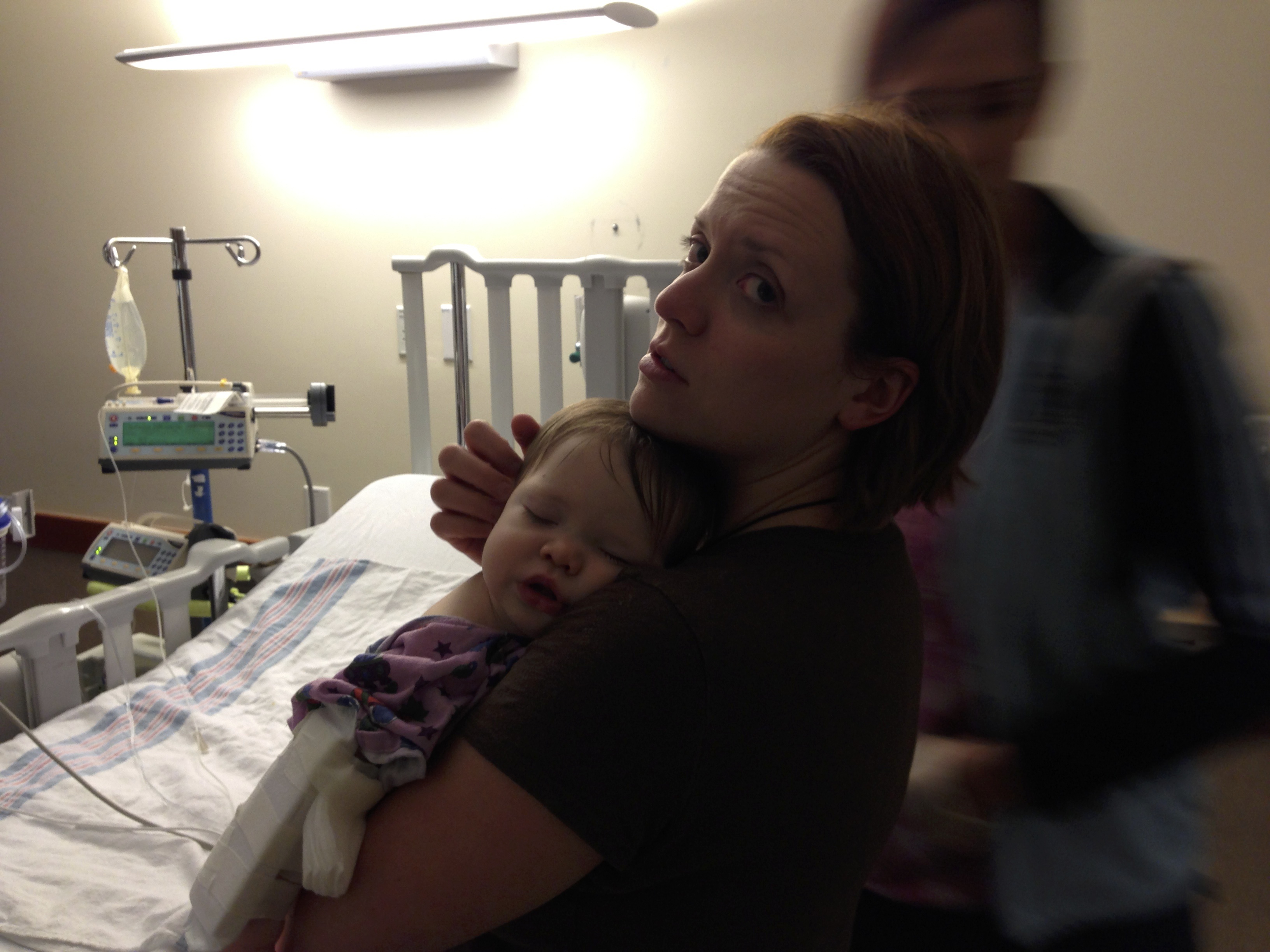
Lucy was in the hospital for 10 days. They were full of ups and downs, but, except for one setback the first time they tried to wean her off of the milrinone, she did well. She responded very well to the medications. They did a battery of tests. Finally, at the very end of her stay, they told us they thought her case was viral, which was good news for her prognosis. They said that children who contracted cardiomyopathy virally, rather than genetically, stood a better chance of recovery. Regardless, 33% of children recover completely, 33% stay stable, and 33% worsen and need a transplant. They said that most cases are idiopathic, meaning they don’t know what caused it. In Lucy’s case, they got a hit on the strain of parvovirus that causes Fifths’ Disease in one of the tests. Somehow, the virus infected her heart instead of just causing a mild illness, as it does with most children.
Lucy went home on four medications. She had to take them all twice a day. We saw the cardiologist once a month. After the New Year, she got Synagis shots (to prevent RSV) once a month at a different pediatrician’s office.
At the first visit, the doctor cautiously predicted that Lucy was on “a trajectory of recovery”. She thought Lucy would be better and be off meds in six months.
At the next visit, a week before Christmas, things didn’t look so optimistic. Lucy wasn’t improving anymore. The doctor still thought we would know which course the disease would take within six months. She was definitely right on that score.
After Christmas, Lucy just wasn’t doing well. I don’t remember the details about how dilated her heart was or what her ejection fraction was. I struggled to keep track of those things while she was sick. But she was not improving. In fact, she seemed to be getting worse instead of better. At the beginning of February, the doctor began talking about the need for a “tune-up” in the hospital. When I asked her if it was normal to need one, she said it wasn’t a good sign that it was needed this soon.
At the end of February, Lucy was once again admitted to the PICU at Dell Children’s. She perked up quickly with the milrinone and the blood transfusions. After five days, they told us she could go home the next day and moved us to the intermediate floor.
We didn’t get moved to the new room and fall asleep until 11 p.m. A few hours later, we were awakened by a resident. I couldn’t comprehend what she was telling us at first. I finally made out that she was saying we had to go back to the PICU right now. Lucy had a run of ventricular tachycardia (according to Wikipedia, “a type of tachycardia, or a rapid heartbeat, that starts in the bottom chambers of the heart”) and did not tolerate it well, according to the nurse who checked her during it. They had to take us back to the PICU and send us to Dallas by helicopter the next morning.
I am embarrassed of this now, but I argued with the doctor. I kept stupidly saying that Lucy needed to sleep. I didn’t want them to wake her up and move her. I was confused about all the worry, since none of us had woken up and Lucy seemed to be sleeping peacefully. The doctor finally gave up trying to convince me and we went back to the PICU.
None of us slept any more that night. Lucy took a brief nap early in the morning. A few hours later, she and I departed for Dallas on a STAR flight and Chris went home to pack for both of us and get Max.
I will never forget that helicopter ride. I could barely stay awake and yet I felt like I was willing Lucy to stay alive every moment. All I did the entire way there was will her to not have another attack of vtach and to stay alive. I was not allowed to ride in back with her, but the paramedics talked to me through my headset and assured me she was doing fine.
We arrived at Parkland and rushed her next door to the Cardiac ICU at Children’s Medical Center of Dallas. Everything happened so fast. That hospital was so different from Dell. I was alone in a strange hospital with my sick daughter and my husband miles away. A woman beckoned me out of the room and I reluctantly left. I didn’t realize she was going to take me all the way off the floor and have me sign some papers. By the time I made it back, they were about to sedate Lucy just for an Echo, because she was so tired and upset. I told them in no uncertain terms that there was no need for that. She was just hungry and would calm down if I gave her a bottle. Then, they could do the Echo. I couldn’t believe they would pull me away from my daughter, leaving her alone, and then make a decision like that without my consent. Over an Echo.
Sure enough, she calmed down and they were able to do the test with no problem. However, they had to do an IV after that.
Poor Lucy had a terrible time with IVs. She was a terrible stick. They always had trouble finding a vein and then getting it in, especially after she had already been in the hospital for awhile and had several IVs already. It caused her so much pain. I had to hold her down and try to comfort her while they did this. Usually, I stayed stoic for her, but on that day, I was too exhausted and frightened. I couldn’t hold it together anymore. I laid my head down next to hers and sobbed with her in front of everyone. After that, they all left us alone and I held her as she fell asleep in my arms. I sat there holding her until Chris arrived soon after.
The next few days were surreal. We simply held our breaths every moment, hoping against hope that she wouldn’t have another episode of ventricular tachycardia. Seconds, minutes, then days, went by without another attack. The doctors were surprised. I was starting to suspect that someone might have overreacted somewhere, but I felt like I wasn’t qualified to have that opinion. Every night I went to sleep, fearing she would die during the night. But I had to let go and sleep and trust everything would be ok. I was simply, fiercely determined that she would not have another attack and she would not die.
During her time in the CICU, we met with the entire transplant team. They interviewed us and performed all of the tests to ensure Lucy was a good candidate for transplant.
They moved us to the regular cardiac floor and we stayed there for 2 ½ weeks. They basically watched her the entire time. There were no medical procedures. They were afraid to let her go home. They were still very worried there would be another attack of vtach and it would prove fatal. She did have several runs of it, but they were all within normal levels. Apparently, anyone can have ventricular tachycardia at anytime and still have a healthy heart. You don’t even know it has happened. You just go on about your day. It was just a worry for Lucy, because of the dilated cardiomyopathy. They were afraid it was a sign that her heart was becoming increasingly irritated. She tolerated each run well, though, and they didn’t happen that often. We didn’t even know they were happening, unless we saw it on the monitor.
A rhythm expert finally came to us and told us she had looked at all of Lucy’s records since her admission to Children’s and she did not think Lucy’s vtach was medically dangerous. This was very different from what the other doctors were saying. We were elated and it jived with what I was thinking. But it seemed too good to be true to go from thinking our daughter would die any minute to the vtach not being a worry at all.
We had a meeting with all of the doctors involved in Lucy’s case and members of the transplant team. It was agreed that Lucy would be listed as Status II for transplant, meaning she could go back to Austin to live. She would go home on a holter monitor. She would see the transplant team in Dallas once a month and have a new holter monitor placed at each of those visits. She would see her cardiologist in Austin once a week. She would also start physical and occupational therapy to help her catch up with her peers, since her condition had delayed her to the point where she couldn’t even crawl. Her medications were basically unchanged, if I remember correctly. I think they tweaked the dosages a bit. They had tried putting her on Sotalol while we were there. Sotalol is an anti-arrhythmia drug, but anti-arrhythmia drugs can cause arrhythmias as well as prevent them. She had to have it three times a day. We woke up to give it to her an hour before her other morning meds were due. Then, we tried to get a little more sleep before the rest of the meds were due. She also had to have it at midday and 10 p.m. We did our best to barely wake her up, give her the med, and get her back to sleep as quickly as possible. Then we had to get ourselves to sleep as quickly as possible before the morning dose of Sotalol was due.
I was completely willing to do this for her, but I can’t pretend I wasn’t relieved when the doctors decided the Sotalol wasn’t necessary.
We took Lucy home on Friday, March 15. We were so happy. I had this overwhelming feeling that everything would be all right. I really thought she would get better, or stay stable, or get a transplant and survive. I was so sure.

Instead, she died almost exactly two months later, on May 12. It was one day after her 15-month birthday. It was three days after my birthday. It was the evening of Mother’s Day. It was six months after her diagnosis.
She seemed more tired that weekend and had vomited once on Saturday and once on Sunday. We were in communication with her cardiologist and the transplant team and had moved up her appointment with her pediatrician to Monday morning. But when I went to get her up from her nap on Sunday evening, she was cold and she wasn’t breathing. I rushed her to my husband in a blind panic. My only thought was that we had to get her breathing right now. Max didn’t understand our panic and rolled around on the floor laughing. I couldn’t get him out of the way and put her on the floor to do CPR, so I handed her to Chris and he tossed me his phone. He performed CPR while I talked to the paramedics. Then, the paramedics worked on her a very long time. But they were unable to bring her back. I knew in my heart they wouldn’t be able to.
Lucy has been gone almost 9 months. We were expecting a baby at the end of February, but he or she was lost to miscarriage last August. I am happy to announce that we are now expecting a little girl at the end of July/beginning of August. We have not gotten a good look at her heart yet, but she gives every appearance of being perfectly healthy. We plan to name her Scarlett Louise.
I still worry every time Max gets sick. I worry that this baby will have cardiomyopathy, too, or something else. Chris finally had an Echo last month and I was terrified at times that he would have it. But he’s fine. We all are. We miss our Lucy Blythe so much, especially as she would have been two-years-old tomorrow. I wanted this birthday so much. I knew it would be a struggle to get here, but I really thought we would.
Cardiomyopathy is a terrible disease. There is no surgical fix. There are no really effective treatments for it. There has been no change in outcome for these children in 30 years. All children’s diseases are severely underfunded for research, but cardiomyopathy and other heart defects and diseases in children suffer from a singular lack of awareness. We need to keep other families from losing their children. We need to help relieve the stress, fear, uncertainly, and pain that CM kids and their families live with every day. I don’t want my daughter’s death to have been in vain.
For more information about pediatric cardiomyopathy, please visit: www.childrenscardiomyopathy.org.
For more information about Lucy, please “like” and share our Facebook page: www.facebook.com/GoTeamLucy
And follow us on Twitter: www.twitter.com/goteamlucy
And check out our video tribute: www.operationforever.com/lucy